

Tireoide
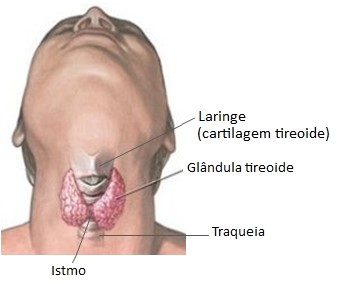
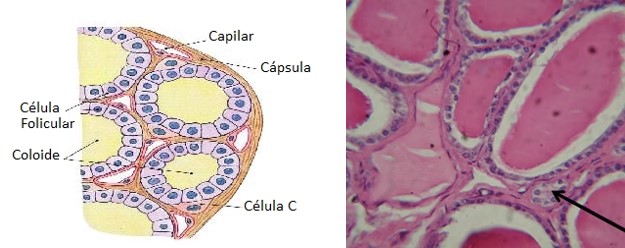
A tireoide é uma glândula endócrina alveolar ou acinosa (porção secretora arredondada), localizada na porção anterior do pescoço, abaixo da laringe e em frente à traqueia, sendo constituída por dois lóbulos unidos por um istmo.
A glândula tireoide é constituída por numerosos folículos esféricos (de 150 a 300 µm de diâmetro), repletos de uma substância secretora, denominada coloide, e revestidos por células epiteliais cúbicas (células foliculares), que secretam seus produtos no interior dos folículos. As células foliculares sintetizam os dois hormônios tireoidianos que contêm iodo, triiodotironina (T3) e tetraiodotironina ou tiroxina (T4). T3 e T4 estão ligados uma glicoproteína, a tireoglobulina (TG), e são armazenados no coloide dos folículos.
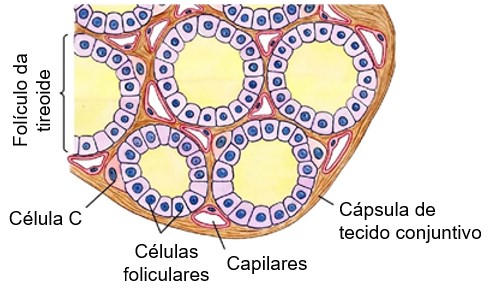
Os folículos são circundados por células parafoliculares, que sintetizam e secretam o hormônio calcitonina. Como consequência, são frequentemente designadas como células C. Localizam-se preferencialmente nas regiões centrais dos lobos da tireoide, onde a atividade das células foliculares é maior e serão tratadas mais abaixo, em um tópico específico.
Hormônios da tireoide
A principal função da tireoide é produzir quantidades adequadas dos dois principais hormônios tireoideanos – T3 e T4 – para atender às demandas periféricas. Esses hormônios controlam a velocidade da maioria das reações químicas intracelulares no organismo, desempenhando importantes papéis na manutenção da homeostasia e regulação do consumo de energia. Seus efeitos fisiológicos, mediados em múltiplos órgãos-alvo, consistem primariamente em estimular o metabolismo e a atividade das células. As funções vitais desses hormônios, particularmente no desenvolvimento, na diferenciação e na maturação, são ressaltadas pelo retardo mental grave observado em lactentes com deficiência da função dos hormônios tireoideanos durante a gestação.
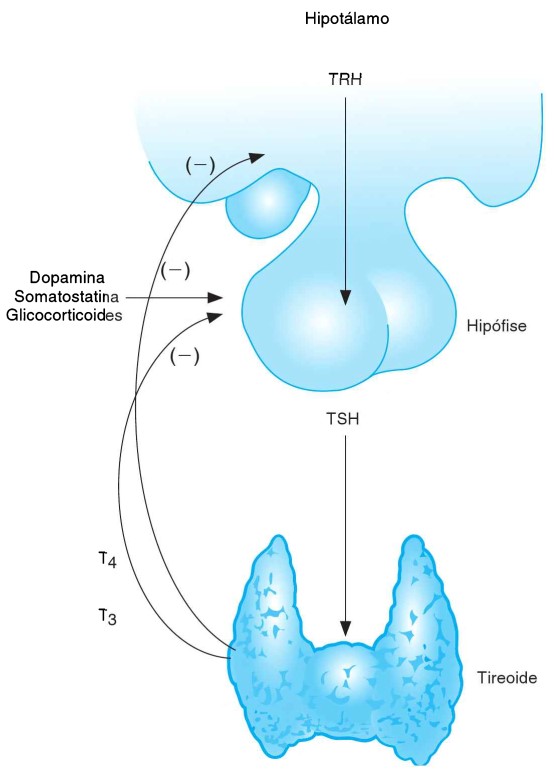
A tireoide é controlada pela atividade do eixo hipotalâmico-hipofisário-tireoidiano. Os hormônios tireoideanos derivam do aminoácido tirosina, sendo produzidos pela glândula tireoide em resposta à estimulação do TSH (hormônio estimulante da tireoide ou hormônio tireoestimulante ou hormônio tireotrópico ou tireotropina). O TSH, produzido pelas células tireotróficas da adenohipófise, liga-se a receptores específicos nas células tireoideanas e estimula todas as etapas síntese do T3 e do T4, bem como sua liberação pela glândula. A síntese e a secreção do TSH, por sua vez, são inibidas pelos hormônios tireoideanos (feedback negativo) e estimuladas pelo hormônio liberador da tireotropina (TRH), produzido no hipotálamo.
Outros fatores que inibem a liberação de TSH incluem os glicocorticoides, a somatostatina e a dopamina.
A síntese dos hormônios da tireoide também é regulada pelo iodo da dieta (obtido na forma de iodeto, primariamente o sal iodado) e envolve as seguintes etapas:
- transporte ativo de iodeto (I–) para o interior da célula tireoidiana;
- oxidação do I– e ligação do iodeto oxidado com resíduos de tirosina da tiroglobulina (TG), formando a monoiodotirosina (MIT) e a diiodotirosina (DIT);
- acoplamento de duas moléculas de DIT para formar o T4, e MIT + DIT para gerar o T3;
- clivagem enzimática da TG, com liberação dos hormônios livres na circulação.
A oxidação do iodo e a reação de acoplamento são catalisadas pela peroxidase tireoideana ou tireoperoxidase (TPO).
Para maiores informações, consulte:
- NUNES, M.T. Hormônios tiroideanos: mecanismo de ação e importância biológica. Arq Bras Endocrinol Metab 2003; 47(6): 639-643.
Disfunções da tireoide
As disfunções da tireoide podem resultar de três fatores:
- alterações nos níveis circulantes dos hormônios tireoideanos;
- comprometimento do metabolismo dos hormônios tireoideanos na periferia;
- resistência às ações dos hormônios tireoideanos em nível tecidual.
O indivíduo cuja função da tireoide está normal encontra-se no denominado estado eutireoideo. O estado clínico resultante da alteração da função da tireoide é classificado em hipotireoidismo (baixa função da tireoide) ou hipertireoidismo (função excessiva da tireoide).
A exemplo da maioria das anormalidades endócrinas, a alteração da função da tiroide pode ser genética ou adquirida, e a sua duração pode ser transitória ou permanente.
As doenças autoimunes desempenham importante papel na doença da tireoide. As respostas imunes anormais dirigidas às proteínas relacionadas com a tireoide resultam em dois processos patogênicos opostos:
- aumento da glândula tireoide (hiperplasia) na doença de Graves,
- destruição da tireoide na tireoidite de Hashimoto.
As principais doenças da glândula tireoide, como a tireoidite (ou doença) de Hashimoto e a doença de Graves, têm uma origem autoimune, ou seja, são provocadas pelo surgimento de anticorpos contra a própria tireoide. Atualmente, conseguimos identificar através de exames de sangue a presença de pelo menos três anticorpos anti-tireoideanos: anti-TPO (anti-tireoperoxidase), TRAb (anti-receptores de TSH) e anti-TG (anti-tireoglobulina).
Para maiores informações, consulte:
- DA FONSECA, C.W.; FLORA ELI MELEK, F.E. Fármacos de amplo uso na prática clínica que interagem com os hormônios tireoidianos. Rev Soc Bras Clin Med 2014; 12(4): 1-7.
Hipotireoidismo
O hipotireoidismo é um distúrbio que resulta de deficiência dos hormônios da tireoide. Tem incidência de 2% nas mulheres adultas e é menos comum nos homens. São reconhecidos dois tipos de hipotireoidismo:
- primário, quando resulta de condições que interfiram diretamente na tireoide (mais frequente),
- central ou secundário, quando resulta de deficiência de TSH.
Cretinismo
O cretinismo é causado por hipotireoidismo extremo em fetos, bebês ou crianças, podendo prolongar-se pela fase escolar, adolescência e idade adulta. Pode ser congênito ou adquirido.
Quando congênito (ocorre in utero), pode resultar da ausência da glândula tireoide ou da incapacidade da tireoide de produzir os hormônios tireoideanos (erro inato do metabolismo). Em ambos os casos, a consequência é o retardo mental grave ou cretinismo, ressaltando o papel vital desempenhado pelos hormônios tireoideanos. O recém-nascido pode até ter aspecto e função normais devido ao suprimento de certa quantidade (geralmente insuficiente) de hormônio tireóideo pela mãe in útero; todavia, dentro de poucas semanas após o nascimento, seus movimentos tornam-se lentos, e tanto o crescimento físico quanto o mental ficam acentuadamente retardados. O crescimento esquelético fica tipicamente mais inibido do que o dos tecidos moles. Como consequência dessa velocidade desproporcional de crescimento, os tecidos moles podem aumentar de modo excessivo, dando à criança a aparência de obesa, pequena e robusta. Em certas ocasiões, a língua fica tão grande, em relação ao crescimento esquelético que impede a deglutição e a respiração, produzindo respiração gutural típica que, por vezes, sufoca o bebê.
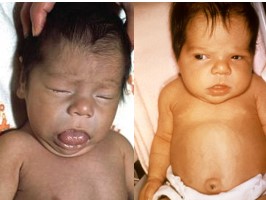
Em geral, o tratamento determina a normalização do crescimento físico; todavia, a não ser que o paciente seja tratado dentro de poucas semanas após o nascimento, o desenvolvimento mental ficará permanentemente retardado.
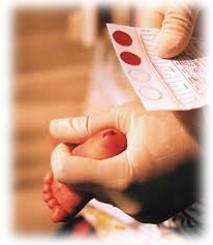
Hoje, toda a criança nascida em território brasileiro tem direito ao Teste do Pezinho Básico, totalmente gratuito pelo SUS. A finalidade do teste é o rastreamento neonatal de crianças portadoras de doenças que devem ser diagnosticadas e tratadas o mais precocemente possível, a fim de evitar sequelas para o paciente. Entre as doenças detectadas pelo teste, o hipotireoidismo congênito está incluído.
Quando o cretinismo é adquirido (cretinismo endêmico), é provocado pela deficiência de iodo na dieta (as necessidades mínimas de iodo não são atingidas no dia a dia). Neste caso, ocorre alteração funcional da tireoide (com queda de T4 sérico e elevação do TSH), além de aumento expressivo da glândula se a carência de iodo permanecer crônica, o que é denominado bócio endêmico. Se não tratado, o retardo mental prolonga-se pela fase escolar, adolescência e idade adulta, levando as crianças a terem baixo rendimento escolar, dificuldade de adaptação social, incapacidade relativa de trabalho na vida adulta e mesmo sérios problemas cognitivos.
Para maiores informações sobre cretinismo, consulte:
- KNOBEL, M.; MEDEIROS-NETO, G. Moléstias associadas à carência crônica de iodo. Arq Bras Endocrinol Metab 2004; 48(1): 53-61.
- MACIEL, L.M.Z.; KIMURA, E.T.; NOGUEIRA, C.R.; MAZETO, G.M.F.S.; MAGALHÃES, P.K.R.; NASCIMENTO, M.L.; NESI-FRANÇA, S.; VIEIRA, S.E. Hipotireoidismo congênito: recomendações do Departamento de Tireoide da Sociedade Brasileira de Endocrinologia e Metabologia. Arq Bras Endocrinol Metab 2013; 57(3): 184-192.
- MINISTÉRIO DA SAÚDE. COORDENAÇÃO GERAL DA POLÍTICA DE ALIMENTAÇÃO E NUTRIÇÃO (CGPAN). Dicas em Saúde: Deficiência de iodo. 2007. Disponível em: <https://bvsms.saude.gov.br/bvs/dicas/68def_iodo.html> acesso em 02/07/2021.
- SOCIEDADE BRASILEIRA DE ENDOCRINOLOGIA E METABOLOGIA. Hipotireoidismo Congênito. Projeto Diretrizes. Associação Médica Brasileira e Conselho Federal de Medicina, 2005. Disponível em: <https://diretrizes.amb.org.br/_BibliotecaAntiga/hipotireoidismo-congenito.pdf> Acesso em 02/07/2021.
Hipotireoidismo em adultos
Nos adultos, o hipotireoidismo pode estar associado a uma diminuição da tireoide devido, por exemplo, à cirurgia ou tratamento com iodo radioativo (usado no manejo do hipertireoidismo da doença de Graves). Entretanto, na maioria das vezes, o hipotireoidismo primário está associado a um aumento da glândula tireoide (bócio) como consequência da deficiência dietética de iodo (bócio coloide endêmico; a falta de iodo impede a produção de T3 e T4, mas não interrompe a formação de tireoglobulina) ou da infiltração linfocítica por uma doença autoimune (tireoidite de Hashimoto, na qual o organismo produz auto-anticorpos e linfócitos T citotóxicos contra a glândula tireoide, levando a uma inflamação crônica – tireoidite – com deterioração progressiva e fibrose da glândula).
 |  |
As manifestações fisiológicas do hipotireoidismo incluem:
- pele seca e escamosa,
- redução do crescimento dos pelos,
- fadiga e cansaço excessivo,
- extrema sonolência,
- atividade muscular lenta,
- frequência e débito cardíacos diminuídos,
- aumento do peso corporal e dificuldade de emagrecer,
- constipação intestinal,
- lentidão mental,
- desenvolvimento de voz rouca e áspera,
- intolerância ao frio.
A expressão total do hipotireoidismo é conhecida como mixedema, pois desenvolve-se em pacientes com ausência quase total da função dos hormônios tireoideanos. Esses casos (casos graves e prolongados ou crônicos), também apresentam aspecto edematoso (inchaço) de todo o corpo, formando "bolsas" sob os olhos.
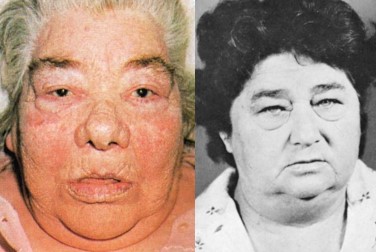
Para maiores informações sobre hipotireoidismo em adultos, consulte:
- ANDRADE, V.A.; GROSS, J.L.; MAIA, A.L. Iodo radioativo no manejo do hipertireoidismo da doença de Graves. Arq Bras Endocrinol Metab 2004; 48(1): 159-165.
- MEDEIROS, C.C.M.; LEMOS-MARINI, S.H.V.L.; BRÍCOLA FILHO, M.; CAMARGO, E.E.; SANTOS, A.O.; MAGNA, L.A; GUERRA JÚNIOR, G.; BAPTISTA, M.T.M.; MACIEL-GUERRA, A.T. Evidências de doença tireóidea auto-imune crônica subclínica em portadoras da Síndrome de Turner. Arq Bras Endocrinol Metab 2007; 51(3): 401-409.
- ROBAZZI, T.C.M.V.; ADAN, F.F. Ocorrência de doenças autoimunes tireoidianas em pacientes com doenças reumáticas. Rev Bras Reumatol 2012; 52(3): 417-430.
- WIERSINGA, W.M. Adult Hypothyroidism. In: FEINGOLD, K.R.; ANAWALT, B.; BOYCE, A. et al. (Editors). Endotext [Internet]. South Dartmouth (MA), MDText.com, 2000. Disponível em: <https://www.ncbi.nlm.nih.gov/books/NBK285561/> Acesso em 02/07/2021.
Hipertireoidismo
Na maioria dos pacientes com hipertireoidismo, toda a glândula tireoide sofre aumento de tamanho de até duas a três vezes o normal; o número de células também aumenta várias vezes mais do que o aumento de tamanho da glândula. Além disso, a velocidade de secreção de cada célula aumenta por várias vezes, chegando até 5 a 15 vezes o normal. Essas alterações da glândula tireoide assemelham-se às produzidas pelo excesso de TSH. Todavia, as concentrações plasmáticas de TSH são inferiores aos valores normais em quase todos os pacientes, quase sempre caindo para praticamente zero.
A doença de Graves, a forma mais comum de hipertireoidismo, é uma doença autoimune, na qual anticorpos tireoestimulantes, designados por TSAb (do inglês, thyroid stimulating antibodies), se ligam na glândula tireoide ao mesmo receptor que liga o TSH, provocando ativação contínua dos sistemas celulares, com resultante desenvolvimento de hipertireoidismo. Esses anticorpos têm efeito estimulante prolongado na secreção da glândula tireoide, durando até 12 horas, em contraste com o curto tempo para o TSH, de pouco mais de 1 hora. O elevado nível de secreção de hormônios tireóideos induzido pelos TSAb suprime, por sua vez, a formação de TSH pela adenohipófise.
As manifestações fisiológicas do hipotireoidismo incluem:
- intolerância ao calor,
- sudorese aumentada,
- perda de peso de leve a extrema, podendo atingir até 37 kg,
- graus variáveis de diarreia,
- fraqueza muscular,
- nervosismo ou outros distúrbios psíquicos,
- fadiga extrema, porém com incapacidade de dormir,
- tremor das mãos,
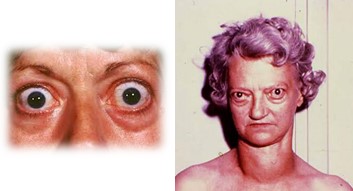
- exoftalmia (protrusão dos globos oculares), podendo se tornar grave (distender o nervo óptico o suficiente para lesar a visão). Com frequência ocorre lesão ocular, pois as pálpebras não conseguem fechar por completo quando a pessoa pisca ou dorme. Em consequência, as superfícies epiteliais dos olhos ficam secas e irritadas e, quase sempre, infectadas, com consequente ulceração da córnea.
Tireoidite de Hashimoto x Doença de Graves
Durante a tireoidite de Hashimoto, os linfócitos T CD4+ auto-reativos recrutam células B e células T CD8+ na tireoide. A progressão da doença leva à morte das células da tireoide e hipotireoidismo. Ambos, os auto-anticorpos e linfócitos T citotóxicos (CLT) contra a glândula tireoide têm sido propostos como responsáveis pela doença.
Na doença de Graves, as células T CD4+ ativadas induzem células B a secretarem anticorpos estimulantes da tireoide contra o receptor do hormônio estimulante da tireoide, resultando em desenfreada produção de hormônio da tireoide e hipertireoidismo.
Para maiores informações, consulte:
- CARDIA, M.S.; KNOBEL, M.; LIMA, N.; GIANELLA-NETTO, M.L.C.C.; CAVALIERE, H.; MEDEIROS-NETO, G. Comparação entre diferentes métodos para avaliar a presença de auto-anticorpos dirigidos ao receptor de TSH em pacientes com moléstia de Graves-Basedow. Arq Bras Endocrinol Metab 2001; 45(6): 563-569.
- STASSI, G.; DE MARIA, R. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat Rev Immunol 2002; 2(3): 195-204.
- VIEIRA, J.G.H.; KASAMATSU, T.S.; HAUACHE, O.M.; MACIEL, R.M.B. Anticorpos anti-tiróide: aspectos metodológicos e importância diagnóstica. Arq Bras Endocrinol Metab 2003; 47(5): 612-621.
Veja também:
- Cansaço, mudanças no peso e mais: 11 sinais de que sua tireoide não vai bem.
- Dieta e tireoide: atenção aos alimentos que podem desregular a glândula.
- Disfunções tireoideanas podem impactar sua libido? Entenda a relação.
- Exame para classificar nódulos de tireoide evita cirurgias desnecessárias.
Células parafoliculares da tireoide (células C) e calcitonina
Como descrito previamente, os folículos da tireoide são circundados por células parafoliculares ou células C (situadas no interstício, entre os folículos da glândula tireoide), que sintetizam e secretam o hormônio calcitonina.
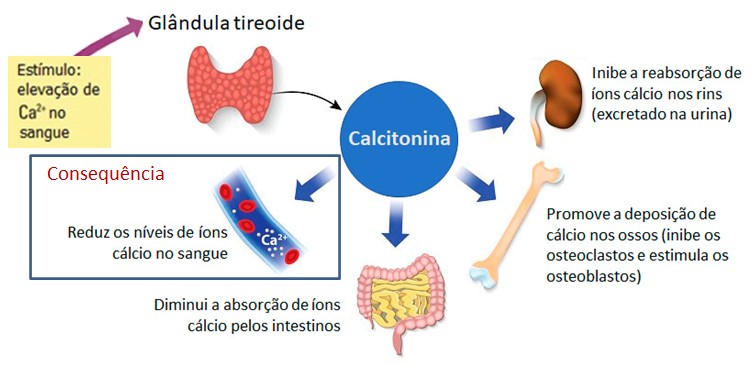
A calcitonina atua na regulação dos níveis plasmáticos de cálcio (Ca2+) e fosfato (HPO42–) e sua secreção independe dos hormônios TSH e TRH, dependendo principalmente do aumento da concentração plasmática de cálcio (Ca2+). As elevações nos níveis plasmáticos de Ca2+ superiores a 9 mg/dL estimulam a liberação de calcitonina. A liberação de calcitonina também é estimulada pela gastrina, um hormônio produzido por células endócrinas da parede do estômago.
O principal tecido-alvo da calcitonina é o tecido ósseo. Quando a concentração sanguínea de Ca2+ está alta, a calcitonina reduz a quantidade de cálcio e de fosfato no sangue, inibindo a reabsorção óssea pelos osteoclastos e acelerando a captação de cálcio e de fosfato pelos osteoblastos e também o tempo de vida destas células. Daí resulta o decréscimo dos níveis sanguíneos de cálcio e de fosfato causado pelo aumento da deposição óssea.
A calcitonina também promove aumento da excreção renal de Ca2+.
A calcitonina, com meia-vida de aproximadamente 5 minutos, é metabolizada e depurada, respectivamente, pelo fígado e pelos rins.
Não existe nenhuma condição patológica associada diretamente com a falta de secreção de calcitonina.
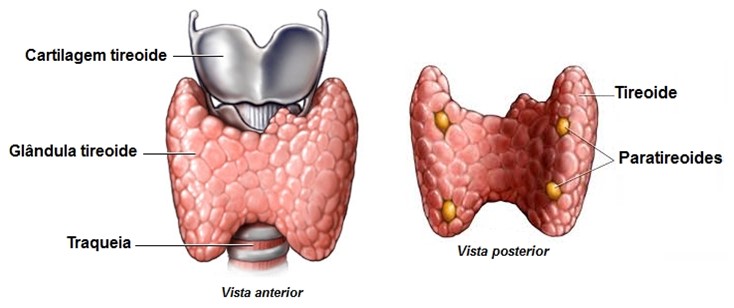
Paratireoides
As paratireoides são quatro pequenas glândulas encaixadas nos polos superiores e inferiores da face dorsal ou posterior da glândula tireoide. Normalmente, uma glândula paratireoide superior e uma inferior estão presas a cada lobo da glândula tireoide. Cada pequena glândula paratireoide mede 3 x 6 mm e possui massa de aproximadamente 40 mg (0,04 g).
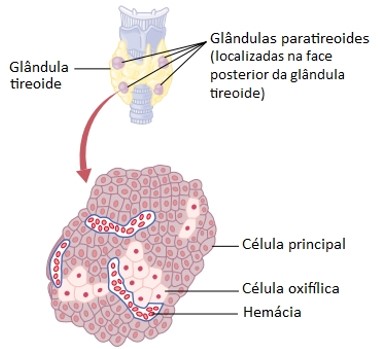
Microscopicamente, as glândulas paratireoides contêm dois tipos de células epiteliais. As células mais numerosas, chamadas de células principais, produzem hormônio paratireóideo (PTH), chamado de paratormônio. A função do outro tipo de célula, chamada de célula oxifílica, é desconhecida.
O hormônio paratireóideo (PTH) é o principal regulador das concentrações sanguíneas dos íons cálcio (Ca2+), magnésio (Mg2+) e fosfato (HPO42–). De maneira semelhante à calcitonina, a liberação do PTH também depende das concentrações plasmáticas de cálcio, porém o PTH é liberado quando a concentração sanguínea de Ca2+ está baixa e tem ação antagônica à da calcitonina. Seus principais órgãos-alvo são os ossos e os rins.
Nos ossos, o PTH estimula o aumento da atividade e da quantidade dos osteoclastos para elevar a concentração do cálcio. O resultado é uma reabsorção óssea elevada, que libera íons cálcio (Ca2+) e fosfato (HPO42–) no sangue. Entretanto, não existem receptores para a PTH nos osteoclastos, mas sim nos osteoblastos e nas células do estroma da medula óssea. Assim, o PTH liga-se aos receptores dos osteoblastos, o que promove um aumento na atividade dos osteoclastos (o PTH promove a mobilização de cálcio ósseo através de estímulo sobre os osteoclastos, via osteoblastos).
Nos rins, o PTH diminui a velocidade na qual Ca2+ e Mg2+ do sangue são eliminados na urina. Também aumenta a excreção de HPO42– na urina. Como mais HPO42– é excretado na urina do que absorvido pelos ossos, o PTH diminui a concentração sanguínea de HPO42– e aumenta as concentrações sanguíneas de Ca2+ e Mg2+.
Um terceiro efeito do PTH nos rins é o de promover a formação do calcitriol, a forma ativa da vitamina D. O calcitriol, também conhecido como 1,25-di-hidroxicolecalciferol [1,25 (OH)2 D], aumenta a velocidade de absorção de Ca2+, HPO42– e Mg2+ pelo intestino, elevando os níveis sanguíneos de cálcio.
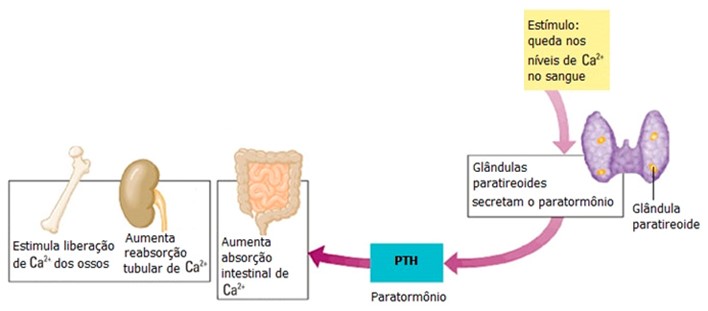
A estreita regulação da liberação de PTH pelos níveis circulantes de cálcio é um exemplo de regulação por retroalimentação (feedback) negativa: o principal efeito fisiológico do PTH consiste em manter a homeostasia do Ca2+ plasmático. Sua liberação é controlada através de um estreito sistema de retroalimentação pelas concentrações plasmáticas de Ca2+. A ocorrência de pequenas alterações nos níveis plasmáticos de Ca2+ é detectada pelo receptor paratireóideo sensor de Ca2+.
Regulação hormonal dos níveis plasmáticos de cálcio
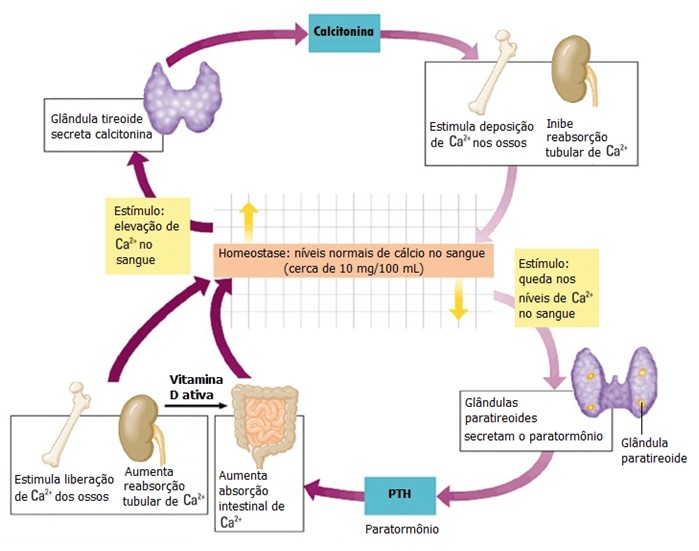
A regulação dos níveis plasmáticos de Ca2+ é decisiva para a função normal das células, transmissão neural, estabilidade das membranas, estrutura óssea, coagulação sanguínea e sinalização intracelular. Essa regulação baseia-se nas interações entre o paratormônio (PTH) das glândulas paratireoides, calcitriol (vitamina D ativa) e a calcitonina da glândula tireoide. Esses hormônios afetam principalmente três órgãos: os ossos, os rins e o intestino delgado.
O principal estímulo para a secreção do PTH pelas paratireoides é o decréscimo dos níveis plasmáticos de cálcio. Já a elevação dos níveis de cálcio no plasma inibe a secreção de PTH e estimula a secreção de calcitonina pelas células C da tireoide. Esta regulação mantém a variação dos níveis de calcemia (níveis de cálcio ionizado no sangue) dentro de limites normais.
O PTH estimula a reabsorção óssea e liberação de Ca2+ na circulação. Nos rins, o PTH promove a reabsorção de Ca2+ e excreção de fosfato inorgânico (Pi) na urina. Além disso, o PTH estimula a produção da forma ativa da vitamina D (calcitriol), a qual aumenta a absorção intestinal do Ca2+ dietético e reabsorção renal do Ca2+. A calcitonina opõe-se aos efeitos do PTH através da inibição da reabsorção óssea, aumento da deposição óssea e aumento da excreção renal de Ca2+. O resultado global das interações entre o PTH, a vitamina D ativa e a calcitonina consiste na manutenção das concentrações plasmáticas normais de Ca2+.
OBS.:
Um aumento simultâneo do Ca2+e do HPO42–conduz à precipitação de fosfato de cálcio nos tecidos moles do organismo, causando a sua irritação e inflamação.
Para maiores informações sobre a vitamina D ativa na regulação hormonal dos níveis plasmáticos de cálcio, veja também:
- DE CASTRO, L.C.G. 2011. O sistema endocrinológico vitamina D. Arq Bras Endocrinol Metab; 55(8): 566-575.
Disfunções das paratireoides
Tanto a hipossecreção como a hipersecreção de PTH desencadeiam sintomas graves.
Hipoparatireoidismo
O hipoparatireoidismo resulta da deficiência ou redução da secreção de PTH abaixo do normal, e pode estar associado a outros distúrbios endócrinos e neoplasias (tumores), ou resultar da remoção cirúrgica das glândulas paratireoides.
Quando as glândulas paratireoides não secretam uma quantidade suficiente de PTH, a reabsorção osteocítica do cálcio plasmático diminui e os osteoclastos tornam-se quase totalmente inativos. Como consequência, a reabsorção de cálcio a partir dos ossos é deprimida a ponto de provocar uma queda nos níveis de Ca2+ nos líquidos corpóreos. Assim, como o cálcio e os fosfatos não estão sendo absorvidos a partir do osso, essa estrutura costuma permanecer resistente.
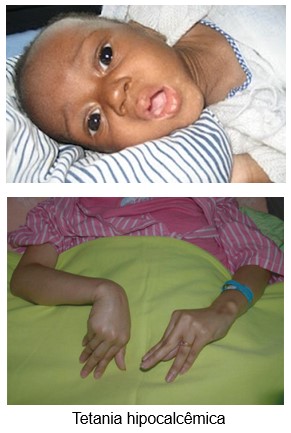
Quando as glândulas paratireoides são subitamente removidas, o nível de cálcio do sangue cai de seu valor normal de 9,4 mg/dL para 6 a 7 mg/dL dentro de 2 a 3 dias (hipocalcemia), enquanto a concentração sanguínea de fosfato pode duplicar. Ao ser alcançado esse baixo valor do cálcio, verifica-se o aparecimento dos sinais habituais de tetania. Dentre os músculos especialmente sensíveis ao espasmo tetânico, destacam-se os músculos da laringe. O espasmo desses músculos obstrui a respiração, constituindo a causa habitual de morte na tetania, a não ser que seja instituído o tratamento adequado.
Os sintomas da hipocalcemia são:
- aumento da excitabilidade,
- espasmos musculares,
- arritmias cardíacas e convulsões,
- tetania hipocalcêmica.
OBS.:
Devido ao importante papel do PTH na regulação aguda dos níveis plasmáticos de Ca2+, a tetania hipocalcêmica constitui manifestação precoce da remoção cirúrgica das glândulas paratireoides.
Para maiores informações, consulte:
- SOCIEDADE BRASILEIRA DE ENDOCRINOLOGIA/SOCIEDADE BRASILEIRA DE CIRURGIA DE CABEÇA E PESCOÇO. Hipoparatireoidismo: diagnóstico e tratamento. Disponível em: <https://diretrizes.amb.org.br/_DIRETRIZES/hipoparatireoidismo_diagnostico_e_tratamento/files/assets/common/downloads/publication.pdf> Acesso em 04/07/2021.
Hiperparatireoidismo
O hiperparatireoidismo resulta da secreção inapropriada e excessiva de PTH. A produção excessiva de PTH é em geral, causada por hiperplasia, adenoma (tumor benigno) ou carcinoma (tumor maligno ou câncer) das glândulas paratireoides (hiperparatireoidismo primário). Tais tumores ocorrem com mais frequência em mulheres, principalmente devido ao estímulo das glândulas paratireoides pela gestação e lactação.
As manifestações clínicas e laboratoriais consistem em:
- níveis elevados de PTH,
- aumento dos níveis plasmáticos de cálcio (hipercalcemia),
- excreção urinária aumentada de cálcio (hipercalciúria) com a formação aumentada de cálculos renais (urolitíase),
- diminuição dos níveis plasmáticos de fosfato,
- aumento da reabsorção óssea pelos osteoclastos (atividade osteoclástica extrema nos ossos),
- aumento de fosfatase alcalina plasmática (secretada pelos osteoblastos que aumentam sua atividade na tentativa vã de produzir quantidade suficiente de novo tecido ósseo para compensar o antigo osso absorvido pela atividade osteoclástica).
Entretanto, tanto o hiperparatireoidismo primário quanto o secundário podem ser assintomáticos (não apresentar sintomas) e muitas vezes os pacientes descobrem o problema através de uma outra condição, como fraturas ósseas provocadas pela extensa descalcificação.
OBS.:
O hiperparatireoidismo secundário ocorre, geralmente, em pacientes com insuficiência renal crônica.
Quando os sintomas do hiperparatireoidismo se manifestam, costumam envolver:
- fadiga/fraqueza,
- dor nos ossos e nas articulações,
- fraturas patológicas,
- cálculo renal,
- náuseas e vômitos,
- dores abdominais.
Além da extensa descalcificação ósseas, as radiografias do osso revelam ocasionalmente, amplas áreas císticas puncionáveis (osteopatia cística), repletas de osteoclastos, os chamados “tumores” osteoclásticos de células gigantes. Além disso, podem ocorrer múltiplas fraturas dos ossos enfraquecidos, quando submetidos aos traumatismos leves, especialmente nos locais de desenvolvimento dos cistos. A osteopatia cística do hiperparatireoidismo recebe o nome de osteíte fibrosa cística.
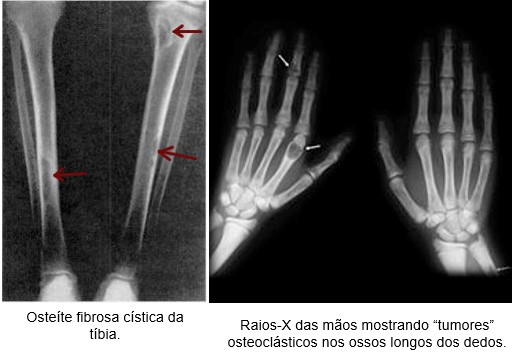
Para maiores informações, consulte:
- BANDEIRA, F.; GRIZ, L.; CHAVES, N.; CARVALHO, N.C.; BORGES, L.M.; LAZARETTI-CASTRO, M.; BORBA, V.; DE CASTRO, L.C.; BORGES, J.L.; BILEZIKIAN, J. Diagnosis and management of primary hyperparathyroidism: a scientific statement from the Department of Bone Metabolism, the Brazilian Society for Endocrinology and Metabolism. Arq Bras Endocrinol Metab 2013; 57(6): 406-424.
Adrenais (suprarrenais)
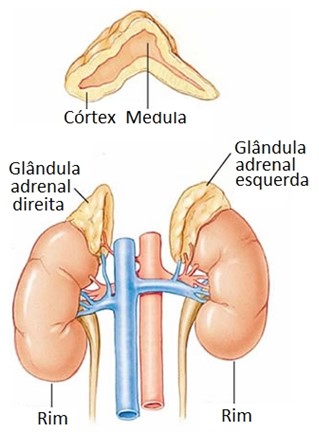
As glândulas adrenais ou suprarrenais são duas glândulas achatadas de formato piramidal e coloração amarela, situadas sobre o polo superior de cada rim. No adulto, cada glândula adrenal mede 3 a 5 cm de altura, 2 a 3 cm de largura, e pouco menos de 1 cm de espessura, com massa de 3,5 a 5 g. Ela é geralmente mais pesada no homem do que na mulher, mas o tamanho pode apresentar ampla variação em função da demanda secretora.
Tal como os rins, as adrenais são órgãos retroperitoneais, estão envolvidas por abundante tecido adiposo, são revestidas por uma cápsula de tecido conjuntivo e providas de uma rede vascular bem desenvolvida.
Estrutural e funcionalmente, cada glândula suprarrenal é composta por duas regiões distintas, o córtex adrenal, região externa de tonalidade amarelada e que compreende a maior parte da glândula (80% a 90% da glândula); e a medula adrenal, região central, marrom-avermelhada e menos volumosa (10% a 20% da glândula).
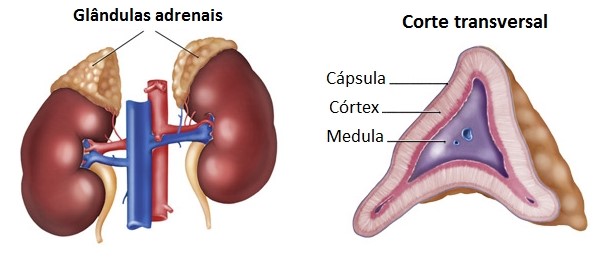
Essas duas regiões podem ser consideradas dois órgãos distintos, de origens embriológicas diferentes, apenas unidos anatomicamente. O córtex é um tecido glandular derivado do mesoderma embrionário, enquanto a medula, mais parecida com um agrupamento de células nervosas do que com uma glândula, tem origem nas células da crista neural, a qual também vai originar os neurônios pós-ganglionares da divisão simpática do sistema nervoso autônomo.
Cada região produz também seu próprio grupo de hormônios. O córtex, de origem mesodérmica, produz hormônios esteroides (derivados do colesterol) chamados de corticosteroides. A medula, de origem neuroectodérmica, é funcionalmente relacionada ao sistema nervoso simpático e secreta as catecolaminas adrenalina (epinefrina) e noradrenalina (norepinefrina) em resposta ao estímulo simpático.
O córtex adrenal
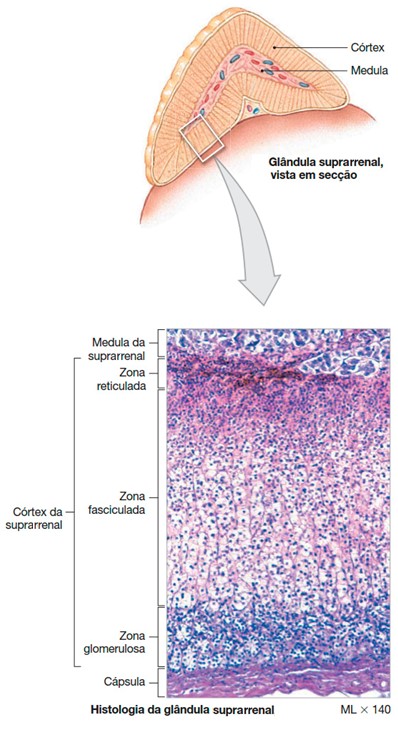
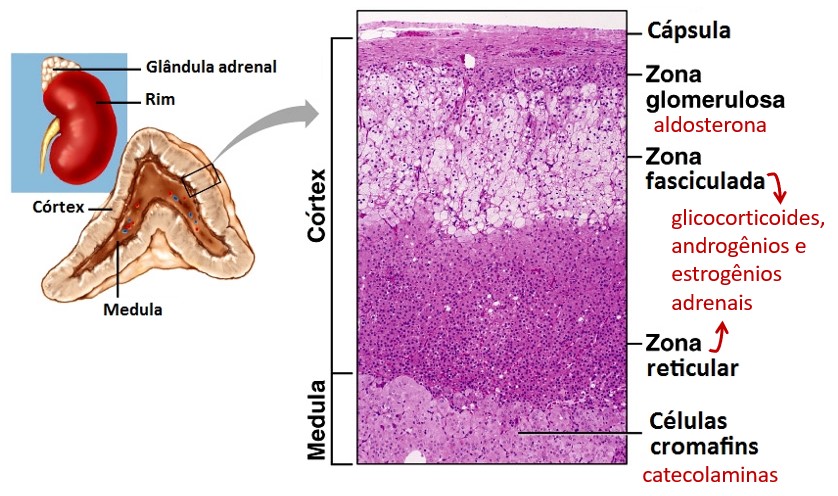
O córtex da suprarrenal é formado por três zonas concêntricas distintas:
- zona glomerulosa: camada externa
- zona fasciculada: camada média
- zona reticulada: camada interna
Estas três zonas são especializadas, tanto funcional como morfologicamente.
A zona glomerulosa, que responde por cerca de 15% do volume cortical, situa-se imediatamente sob a cápsula e é composta por pequenos aglomerados celulares.
Sob a zona glomerulosa encontra-se a porção mais densa do córtex suprarrenal, a zona fasciculada, que representa aproximadamente 78% do volume cortical. Nesta camada, as células formam longas colunas ou feixes que se estendem da superfície para a medula da glândula.
A camada mais profunda do córtex é a zona reticulada, uma estreita camada de cordões celulares organizados irregularmente, que representa apenas aproximadamente 7% do volume celular do córtex da suprarrenal.
A medula adrenal
A medula da suprarrenal contém células secretoras denominadas células cromafins, cromafinas ou feocromócitos. São assim denominadas por causa da sua capacidade de adquirir coloração castanha quando expostas a uma solução aquosa de dicromato de potássio. Essa reação se deve à oxidação das catecolaminas por sais de cromo, produzindo um pigmento castanho.
As células cromafins são células arredondadas e grandes da medula, semelhantes aos neurônios dos gânglios simpáticos (são neurônios pós-ganglionares simpáticos modificados; sem prolongamentos). Essas células são inervadas por fibras pré-ganglionares simpáticas; a ativação simpática fornecida pelos nervos esplâncnicos deflagra a atividade secretora desses neurônios ganglionares modificados.
Hormônios adrenais
Corticosteroides
Os corticosteroides são hormônios esteroides derivados do colesterol secretados pelo córtex adrenal. Podem ser divididos em três grupos, de acordo com suas ações fisiológicas principais:
- mineralocorticoides,
- glicocorticoides,
- androgênios, estrogênios e progesterona (hormônios sexuais).
A zona glomerulosa secreta o principal mineralocorticoide, a aldosterona, importante hormônio que contribui para manter o equilíbrio de sódio, potássio e de água no organismo e, consequentemente, dos níveis da pressão arterial. Sua secreção independe dos hormônios CRH (hormônio liberador de corticotropina) e ACTH (corticotropina ou hormônio adrenocorticotrópico) do eixo hipotalâmico-hipofisário, e está associada à ativação do sistema renina-angiotensina-aldosterona (SRAA) (ver Regulação endócrina da reabsorção tubular).
Os glicocorticoides, dentre os quais um dos mais importantes é o cortisol, são secretados principalmente pelas células da zona fasciculada. A secreção dos glicocorticoides é controlada pelo eixo hipotalâmico-hipofisário através dos hormônios CRH e ACTH. Os glicocorticoides regulam o metabolismo de carboidratos, proteínas e lipídeos, exercendo ações no organismo inteiro. Os glicocorticoides também suprimem a resposta imune. O sistema de defesa do organismo e o córtex adrenal estão, portanto, associados porque o cortisol tem propriedades anti-inflamatórias por meio dos leucócitos (reduz a migração dos leucócitos para a área inflamada e a fagocitose das células lesadas), supressão de citocinas e também ação imunossupressora. Alguns glicocorticoides também apresentam atividade mineralocorticoide, porém de maneira mais fraca que a aldosterona. A zona fasciculada também secreta pequenas quantidades de androgênios e estrogênios adrenais.
A zona reticulada produz secreta os androgênios adrenais desidroepiandrosterona (DHEA) e androstenediona (principalmente DHEA) e também uma pequena quantidade de estrogênios e progesterona e, em menor grau, mineralocorticoides. Em condições normais, o efeito dos androgênios é fraco em humanos. Porém são importantes no desenvolvimento dos órgãos sexuais masculinos na infância e também exercem efeitos leves em mulheres ao longo da vida, como, por exemplo, o crescimento de pelos púbicos e axilares, sendo convertidos em testosterona nos tecidos extrarrenais.
A via de síntese dos hormônios corticosteroides sintetizados pelo córtex adrenal é complexa, possuindo várias etapas e envolvendo diferentes intermediários, dependendo do hormônio que está sendo formado.

Qualquer deficiência na via das reações enzimáticas que levam à síntese dos mineralocorticoides, glicocorticoides e androgênios provoca uma grave patologia. A gravidade das manifestações inclui desde a morte intrauterina até anormalidades que só se tornam evidentes na vida adulta e não são potencialmente fatais. Um defeito enzimático da 21-hidroxilase é responsável por 95% das anormalidades genéticas que ocorrem na síntese dos hormônios esteroides da suprarrenal.
Catecolaminas
A medula da suprarrenal contém duas populações de células endócrinas (células cromafins): uma delas secreta adrenalina (epinefrina), enquanto a outra, noradrenalina (norepinefrina), em resposta direta ao estímulo simpático (a secreção de adrenalina é aproximadamente três vezes maior do que a de noradrenalina). Essas catecolaminas são armazenadas em grânulos de secreção e sua secreção deflagra a utilização da energia celular e a mobilização de reservas energéticas, permitindo ao organismo responder a situações de estresse, preparando-o para a ação, que pode ser de “luta ou fuga”.
Disfunções das adrenais
A hipofunção ou insuficiência adrenal decorre da deficiência de produção hormonal pelas glândulas suprarrenais, podendo ser primária, em que há disfunção ou destruição do córtex da adrenal (como na doença de Addison), ou secundária, decorrente da falta de estímulo adrenal por deficiência da produção de corticotropina (ACTH) pela hipófise ou do hormônio liberador de corticotropina (CRH) pelo hipotálamo (alguns especialistas se referem à disfunção hipotalâmica como terciária).
A hiperfunção adrenal causa síndromes clínicas distintas, dependendo do hormônio envolvido:
- a hipersecreção de aldosterona pelas células da zona glomerulosa do córtex adrenal resulta em hiperaldosteronismo primário (síndrome de Conn);
- a hipersecreção de glicocorticoides pelo córtex adrenal resulta em síndrome de Cushing;
- a hipersecreção de andrógenos pelo córtex adrenal resulta em virilismo adrenal (síndrome adrenogenital);
- a hipersecreção de adrenalina/noradrenalina pela medula adrenal resulta em feocromocitoma;
Essas síndromes, com frequência, têm características que se sobrepõem.
Anormalidades da secreção adrenocortical
Hipoadrenalismo – Doença de Addison
A doença de Addison resulta de níveis anormalmente baixos de aldosterona e de cortisol. A causa de muitos dos casos de doença de Addison é desconhecida (idiopática), mas o desenvolvimento incompleto do córtex adrenal, sua destruição por uma doença autoimune, por infecção severa (p. ex., tuberculose) ou por neoplasia, ou ainda hemorragia suprarrenal estão entre as causas conhecidas. Também pode ser causada por supressão da função hipofisária decorrente de tratamentos prolongados com glicocorticoides ou à destruição do hipotálamo por células neoplásicas.
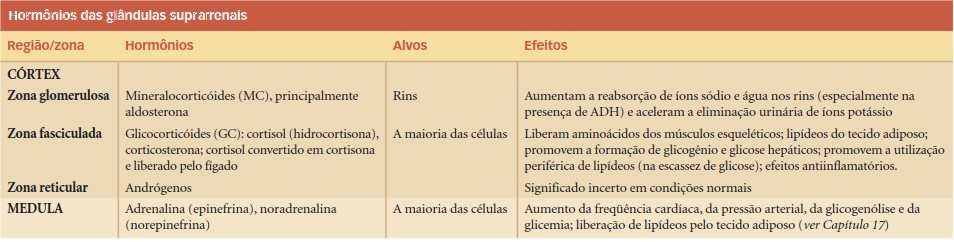
Os hormônios corticosteroides insuficientes resultam em níveis elevados de ACTH hipofisário e, por isso, ocorre uma pigmentação anormal da pele e de áreas da mucosa oral. A insuficiência de cortisol também está relacionada com fraqueza muscular e fadiga. Níveis inadequados de aldosterona interferem no equilíbrio de líquidos e eletrólitos renais, consequentemente reduzindo a pressão sanguínea sistêmica (hipotensão arterial) e contribuindo para um choque circulatório, manifestação particularmente crítica e que requer tratamento imediato.
Os sintomas da doença de Addison compreendem:
- astenia
- fadiga
- perda de peso
- anorexia
- pigmentação anormal da pele e de áreas da mucosa oral
- baixos níveis de Na+, elevados níveis de K+ e redução do pH sanguíneo hipotensão arterial
Para maiores informações sobre doença de Addison autoimune, consulte:
- SILVA, R.C.; KATER, C.E. Doença de Addison de etiologia auto-imune. Arq Bras Endocrinol Metab 1998; 42(6):431-443.
Hiperaldosteronismo primário – Síndrome de Conn
O hiperaldosteronismo (ou aldosteronismo) primário (síndrome de Conn) é causado pela produção excessiva de aldosterona, decorrente de hiperplasia (aumento do número de células) ou neoplasia (tumor) no córtex suprarrenal. Além da secreção inapropriada e excessiva de aldosterona, há supressão da renina plasmática.
Os principais sintomas incluem:
- níveis reduzidos de K+ ( hipocalemia ou hipopotassemia)
- elevação do pH sanguíneo
- retenção de água e de Na+ pelos rins hipertensão arterial
Os pacientes podem apresentar-se assintomáticos, com sintomas decorrentes da crise hipertensiva (cefaleia, tonturas, náuseas, vómitos, dispneia, dor torácica, défices neurológicos e síncope ou desmaio) ou das complicações geradas pela hipocalemia (poliúria, nictúria, cãibras musculares, fraqueza muscular excessiva, parestesias, tetania e até paralisias musculares).
Do ponto de vista laboratorial, além da hipocalemia, os resultados revelam concentrações elevadas de aldosterona plasmática e urinária, concentração de renina plasmática baixa, relação aldosterona/renina elevada e, por vezes, alcalose metabólica.
OBS.:
O hiperaldosteronismo secundário ocorre quando alguns fatores externos, como a produção excessiva de renina, aumentam a secreção de aldosterona.
Para maiores informações sobre hiperaldosteronismo primário, consulte:
- KATER, C.E. Hiperaldosteronismo Primário. Arq Bras Endocrinol Metab 2002; 46(1):106-115.
- KATER, C.E. Hiperaldosteronismo primário: novas tendências. Rev Bras Hipertens 2002; 9:165-173.
- MATOS, F.C. Hiperaldosteronismo primário – uma causa de hipertensão arterial. Rev Port Clin Geral 2008; 24(6): 693-701.
- MIZZACI, C.C.; FERNANDES, R.M.; GALDINO JUNIOR, A.; PAULO OLIVEIRA CARDOSO, P.O.; DULTRA, L.V.; PASSARELLI JR., O.; GONZAGA, C.; BORELLI, F.A.O.; SOUZA, M.G.; CORDEIRO, A.; AMODEO, C. Hiperaldosteronismo primário: somente o tratamento clínico é suficiente? Rev Bras Hipertens 2011; 18(1): 36-40.
- PARMAR, M.S.; SINGH, S. Conn Syndrome. Disponível em: <https://www.ncbi.nlm.nih.gov/books/NBK459197/> Acesso em 06/07/2021.
Hiperadrenalismo – Síndrome de Cushing
A síndrome de Cushing é caracterizada pela hipersecreção de cortisol (hipercortisolismo) e androgênios e, possivelmente, pelo excesso de produção de aldosterona pelo córtex adrenal.
O excesso de glicocorticoides (hipercortisolismo) pode ser decorrente da superprodução por tumor suprarrenal ou estimulação excessiva da síntese e liberação de glicocorticoides da suprarrenal pelo ACTH produzido por tumor hipofisário ou por tumor ectópico (tumor não hipofisário, que geralmente provem de um determinado tipo de tumor do pulmão).
Assim, o hipercortisolismo, conhecido como síndrome de Cushing, pode ser dividido em duas categorias, dependendo de sua etiologia:
- síndrome de Cushing de origem endógena ou independente de corticotropina (ACTH-independente),
- síndrome de Cushing de origem hipofisária ou CTH-dependente (doença de Cushing); maioria dos casos.
Na síndrome de Cushing independente de corticotropina (de origem endógena), a produção excessiva de cortisol resulta geralmente, de um tumor funcional no tecido adrenocortical (adenoma ou carcinoma adrenal), independentemente da estimulação pelo ACTH. A exposição crônica ao excesso de cortisol circulante suprime os níveis de CRH e ACTH no plasma.
Na síndrome de Cushing dependente de corticotropina (de origem hipofisária), os níveis elevados de glicocorticoides são devidos à estimulação excessiva pelo ACTH. A elevação crônica de ACTH provoca o aumento bilateral (hiperplasia) do córtex da suprarrenal. A produção de corticotropina também pode ser ectópica (derivada de tecido extra-hipofisário), mais frequentemente por carcinoma do pulmão das células pequenas. A secreção tumoral de corticotropina não costuma ser suprimida pelos glicocorticoides (perda do feedback negativo), aspecto útil para distinguir a fonte de corticotropina.
O termo doença de Cushing é reservado à síndrome de Cushing causada pela secreção excessiva de corticotropina por tumores de corticotrofos hipofisários e constitui a forma mais comum da síndrome. O hipercortisolismo dependente de ACTH acomete cerca de 80% dos casos da síndrome de Cushing; destes, 85% são síndrome de Cushing e 15% apresentam tumor ectópico produtor de ACTH.
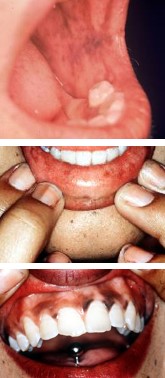
Os sintomas da síndrome de Cushing refletem as múltiplas ações dos glicocorticoides, em particular sobre o metabolismo de carboidratos, proteínas e lipídeos, e incluem:
- ganho de peso (ganho ponderal),
- acúmulo de tecido adiposo na face, conferindo o aspecto típico de face redonda ou face de lua cheia,
- acúmulo de tecido adiposo no tronco (aumento do coxim adiposo dorso-cervical e aumento da gordura que sobressai acima da fossa supraclavicular),
- intolerância à glicose e elevação dos níveis de glicemia,
- hipertensão arterial,
- debilidade e fraqueza musculares (dificuldade de subir escadas ou se levantar de uma cadeira baixa),
- tecidos subcutâneos frágeis com grandes estrias,
- diminuição ou ausência do fluxo menstrual em mulheres pré-menopáusicas,
- diminuição da libido nos homens,
- supressão do sistema imune,
- osteoporose (devido à redução da deposição de proteínas nos ossos).
Nas crianças e adolescentes, o excesso de glicocorticoides causa a interrupção do crescimento linear e ganho ponderal excessivo. Os outros sintomas são frequentemente acompanhados de depressão e insônia. Os pacientes idosos e aqueles com síndrome de Cushing crônica tendem a apresentar afinamento da pele e osteoporose, com dor lombar e colapso vertebral.
Para maiores informações sobre síndrome de Cushing, consulte:
- FREIRE, D.S. Síndrome de Cushing. Disponível em: <https://www.medicinanet.com.br/conteudos/revisoes/1696/sindrome_de_cushing.htm> Acesso em 06/07/2021.
- SOCIEDADE BRASILEIRA DE ENDOCRINOLOGIA E METABOLOGIA/COLÉGIO BRASILEIRO DE RADIOLOGIA. Síndrome de Cushing Independente do Hormônio Adrenocorticotrófico (ACTH). Projeto Diretrizes, 2008. Disponível em: <https://diretrizes.amb.org.br/_BibliotecaAntiga/sindrome-de-cushing-independente-do-hormonio-adrenocorticotrofico.pdf> Acesso em 06/07/2021.
Virilismo adrenal – Síndrome adrenogenital
A síndrome adrenogenital (virilismo adrenal ou hiperandrogenismo) é um conjunto de manifestações clínicas derivadas de uma secreção excessiva de androgênios, geralmente por um tumor adrenocortical secretor de androgênio ou hiperplasia adrenal.
A hiperplasia adrenal é geralmente geralmente congênita (doença metabólica hereditária ou erro inato do metabolismo), sendo causada pela deficiência de uma dessas enzimas da via de síntese dos hormônios corticosteroides:
- 21-hidroxilase (mais comum),
- 11-hidroxilase,
- 3- hidroxiesteroide desidrogenase.
Os efeitos masculinizantes por todo o corpo, mais visíveis em mulheres, dependem do sexo e da idade do paciente no início da manifestação.
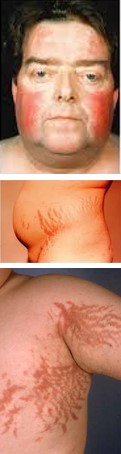
Em neonatos do sexo feminino:
- virilização dos órgãos genitais externos (genitália ambígua).
Em crianças pré-púberes:
- pode ocorrer aceleração do crescimento, mas se não forem tratadas, ocorre fechamento precoce das epífises e baixa estatura;
- nos meninos, ocorre desenvolvimento precoce das características sexuais secundárias e maturação sexual prematura;
- virilização semelhante à que ocorre em mulheres.
Em mulheres adultas: virilização
- hirsutismo (crescimento de barba e distribuição masculina dos pelos corporais e púbicos);
- engrossamento da voz;
- calvície (alopécia) tipo androgênica;
- acne;
- seborreia;
- hipertrofia do clitóris (assemelhando-se a um pequeno pênis);
- deposição de proteínas nos músculos aumento da massa muscular;
- amenorreia;
- atrofia do útero;
- aumento da libido.
Em homens adultos:
- infertilidade.
A deficiência de 21-hidroxilase ainda acompanha distúrbios eletrolíticos em 2/3 dos pacientes, havendo grave perda de sal e morte se não tratados.
Para maiores informações sobre virilismo adrenal, consulte:
- CORRALES HERNÁNDEZ, J.J. Protocolo diagnóstico del hiperandrogenismo adrenal. Hirsutismo. Medicine - Programa de Formación Médica Continuada Acreditado 2008; 10(15): 1018-1020.
- GROSSMAN, A.B. Virilismo adrenal (Síndrome adrenogenital). Disponível em: <https://www.msdmanuals.com/pt-br/profissional/dist%C3%BArbios-end%C3%B3crinos-e-metab%C3%B3licos/dist%C3%BArbios-adrenais/virilismo-adrenal> Acesso em 06/07/2021.
- LANIA, M.V.; ZORRÓN, R.; RAMINHO, M.P.; MOTTA, C.E.T.A.; PINTO, F.E.S.; MARCOS FILGUEIRAS, M. Síndrome Adrenogenital: Tratamento videoendoscópico retroperitoneal em tumor virilizante. Rev Bras Videocir 2006; 4(3): 113-117.
- POLISSENI, F.; GONÇALVES JÚNIOR, H.; VIDAL, V.R.; MACEDO, F.L.; LINS, B.D.; CAMPOS, J.D.; MATTOS, N.B. Síndrome hiperandrogênica em mulher na pós-menopausa: relato de caso. RevBras Ginecol Obstet 2011; 33(8): ; 214-220.
Anormalidades da secreção adrenomedular
Feocromocitoma
Os feocromocitomas são tumores compostos de células cromafins que sintetizam e liberam catecolaminas. Os portadores dessa neoplasia apresentam sintomas clássicos de atividade descontrolada do sistema nervoso simpático, decorrentes da secreção de catecolaminas:
- cefaleia,
- nervosismo intenso,
- sudorese,
- frequência cardíaca elevada (taquicardia),
- palpitações,
- hipertensão arterial (sustentada ou intermitente),
- hiperglicemia,
- aumento na taxa metabólica.
Esses sintomas são comuns a diversas comorbidades, o que dificulta o diagnóstico precoce. Destaca-se a importância dos exames bioquímicos (presença de grande quantidade de ácido vanilmandélico – VMA – na urina, por exemplo) e de imagem na confirmação e localização do tumor.
OBS.:
O ácido vanilmandélico (VMA) é o metabólito final comum das catecolaminas adrenalina e noradrenalina. Sua excreção urinária está aumentada em portadores de feocromocitoma, ganglioneuroma e neuroblastoma.
Para maiores informações sobre feocromocitoma, consulte:
- DOS SANTOS, D.R.P.; BARBISAN, C.C.; MARCELLINI, C.; DOS SANTOS, R.M.V.R. Feocromocitoma e gravidez: Relato de caso e revisão atualizada. J Bras Nefrol 2015; 37(4): 496-500.
- MALACHIAS, M.V.V. Feocromocitoma – diagnóstico e tratamento. Rev Bras Hipertens 2002; 9: 160-164.
- RAMOS, J.A.; PEDRELLI, R.G.R.; FERRAZZA; M.H.S.H.; BORGMANN, G.; KATHERINE PLAUTZ, K. Feocromocitoma: relato de caso. Rev Bras Anal Clin (RBAC) 2020; 52(3):395-399.